INTRODUCTION
Aceruloplasminemia (ACP) is a rare autosomal recessive disorder that ends in the absence or dys-function of ceruloplasmin (CP) [1]. CP is a plasma ferroxidase containing copper and is linked with the oxidation of Fe2+ to Fe3+, allowing it to be transported by transferrin [1]. Its deficiency weakens iron efflux from the tissue and leads to marked iron overload in visceral organs and the central nervous system.
Clinically, the disease has been characterized by the triad of diabetes mellitus, retinal degeneration, and neurological symptoms [2]. The natural history of ACP is primarily unknown, but it is considered progressive and gradually worsens neurological symptoms.
We described a patient presenting with type 2 diabetes and abnormal iron studies due to ACP with typical neuroradiologic abnormality.
CASE REPORT
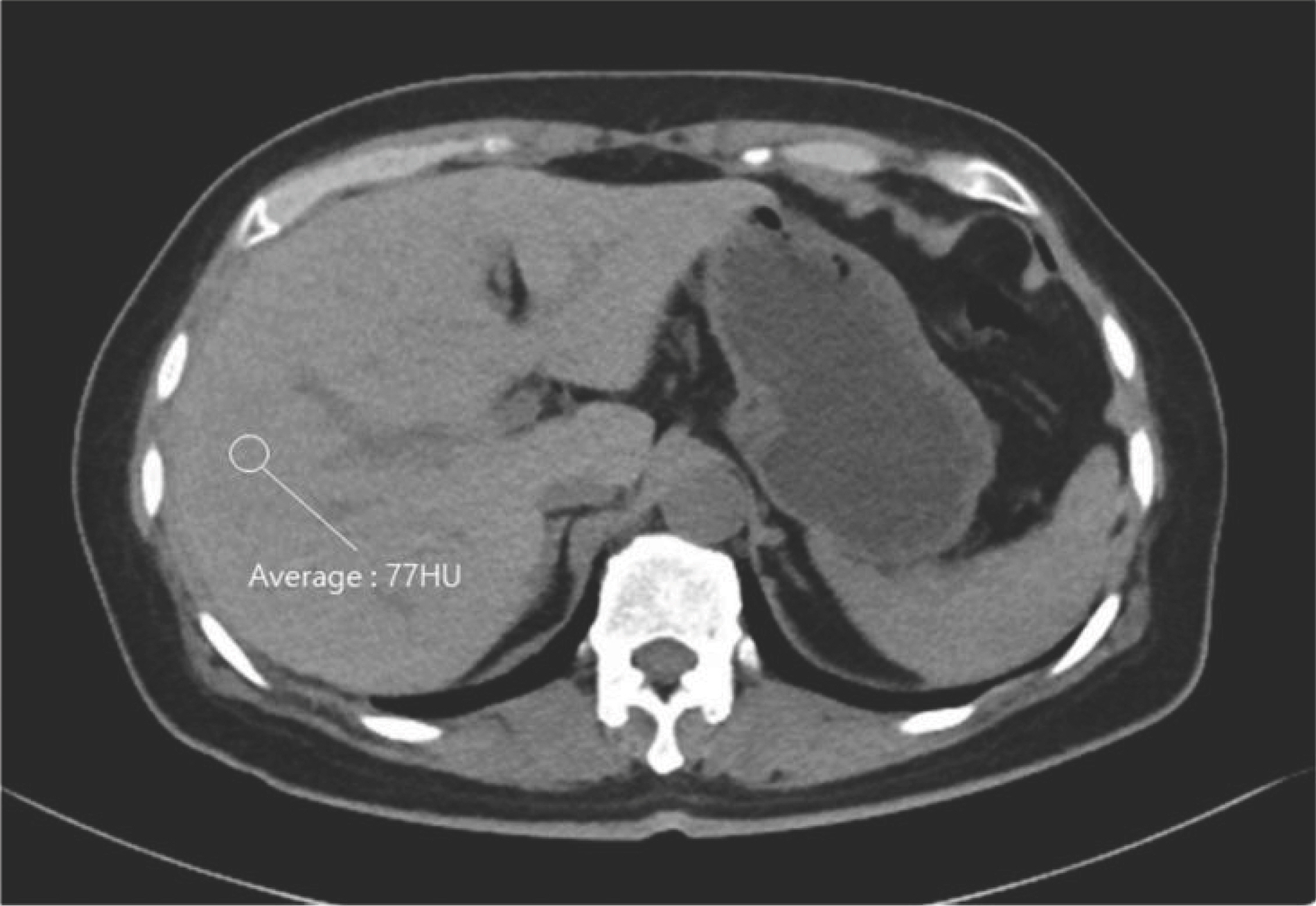
A 56-year-old female patient was admitted to our hospital because of weight loss (about 4 kg over 2 months) with anxiety. Her past medical history in-cluded an 8-year history of hypertension and dyslip-idemia, both compensated and treated with losartan and rosuvastatin, respectively. Body temperature was 36.7Ōäā, blood pressure was 100/70 mm Hg, and heart rate was 76 beats/minute. Height and weight were 146 cm and 52.3 kg. Body mass index was 24.6 kg/m2. She drank alcohol rarely, and there was a positive family history of diabetes mellitus in her two elder brothers. Laboratory tests showed that her fasting plasma glucose level was 170 mg/dL and her HbA1c was 11.9% (reference range, 4.3~5.8%). Fasting C-peptide levels were 1.47 ng/mL (normal range, 0.5~3.5 ng/mL) and her glutamic acid decarboxylase (GAD) antibody was negative (< 0.7 U/mL). Insulin level was not measured at presentation. Kidney, liver, and thyroid function tests were within the normal distribution. She was diagnosed with type 2 diabetes. Ophthalmologic ex-amination with optical coherence tomography and fluorescein angiography revealed branch retinal vein occlusion with small intraretinal hemorrhage from superior arcade but no visual symptoms. She was treated with diet modification and oral hypoglycemic agents (gliclazide, vildagliptin, and metformin), and her glycemic control had become progressively bet-ter. On this admission, initial blood tests revealed he-moglobin 9.2 g/dL (normal range, 12~16 g/dL), red cell distribution width 17.8% (normal range, 12.2~14.8%) and mean corpuscular volume 79.4 fL (normal range, 79~100 fL). She underwent a fecal occult blood test and systematic upper and lower gastrointestinal endoscopy procedures for iron deficiency anemia exploration. There was no evidence of pathological conditions of the gastrointestinal tract associated with iron deficiency. Serum iron content was 15 ╬╝ g/dL (normal range, 29~164 ╬╝ g/dL); transferrin was 237.66 mg/dL (normal range, 200~360 mg/dL); transferrin saturation (TSAT) was 5.245% (normal range, 12~45%) and ferritin was > 1,503 ng/mL (normal range, 10~204 ng/mL). Serum protein electrophoresis and hemolysis screen were normal. C-reactive protein was normal (0.05 mg/dL). She had an abdomen computed tomography (CT) to rule out hidden malignancy as the cause of unintentional weight loss. Abdominal CT showed fat infiltration of the head of the pancreas and high absorbance in the liver (Fig. 1).
Fig.┬Ā1.
Abdominal computed tomography (CT). Unenhanced CT image shows placement of region-of-interest over right hepatic lobe. Liver attenuation is elevated at 77 HU (Hounsfield unit).
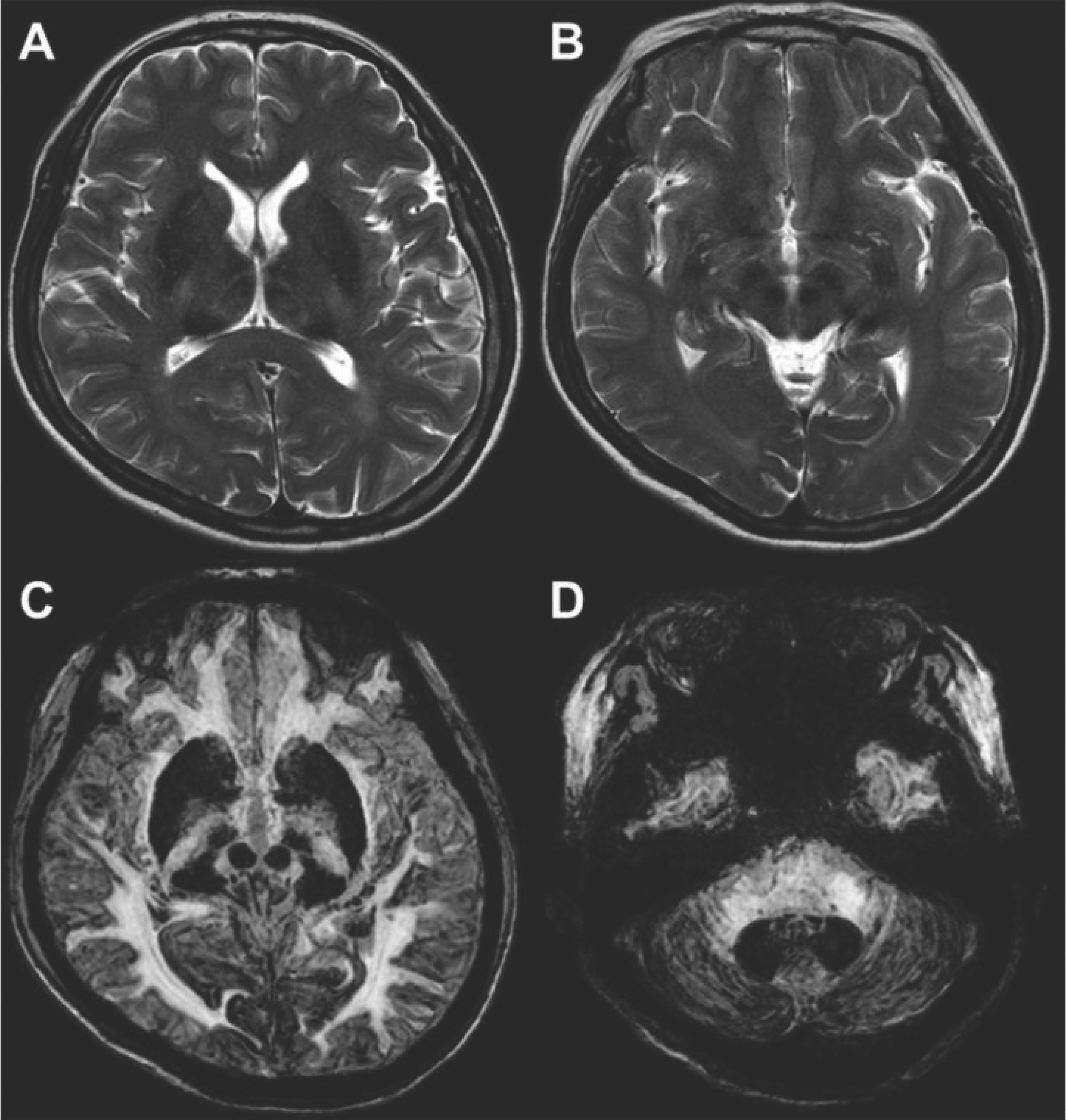
During admission, she was worried about her memory abilities. However, her memory loss was simple and fairly innocuous, such as forgetting to return a phone call. Even though her forgetfulness might have resulted from depression and anxiety of illness, we performed magnetic resonance imaging (MRI) of her brain to rule out organic brain disease. T2-weighted brain MRI showed bilateral symmetric homogenous hypointensity in the putamen, globus pallidus (GP), thalamus, substantia nigra (SN), red nucleus, and dentate nucleus (Fig. 2A, B). Moreover, susceptibil-ity-weighted imaging (SWI) sequences strikingly showed dark signal areas as hypointensity lesions in T2-weighted brain MRI and bilaterally diffuse dark signal along the cerebral cortex and cerebellar folia (Fig. 2C, D), consistent with iron accumulation.
Her speech was almost fluent, and she had no involuntary movement. There was no Kayser-Fleischer ring. There was no atrophy of the limb muscles, and her muscle power was standard. Deep tendon reflex-es were not detected, and the sensation was normal.
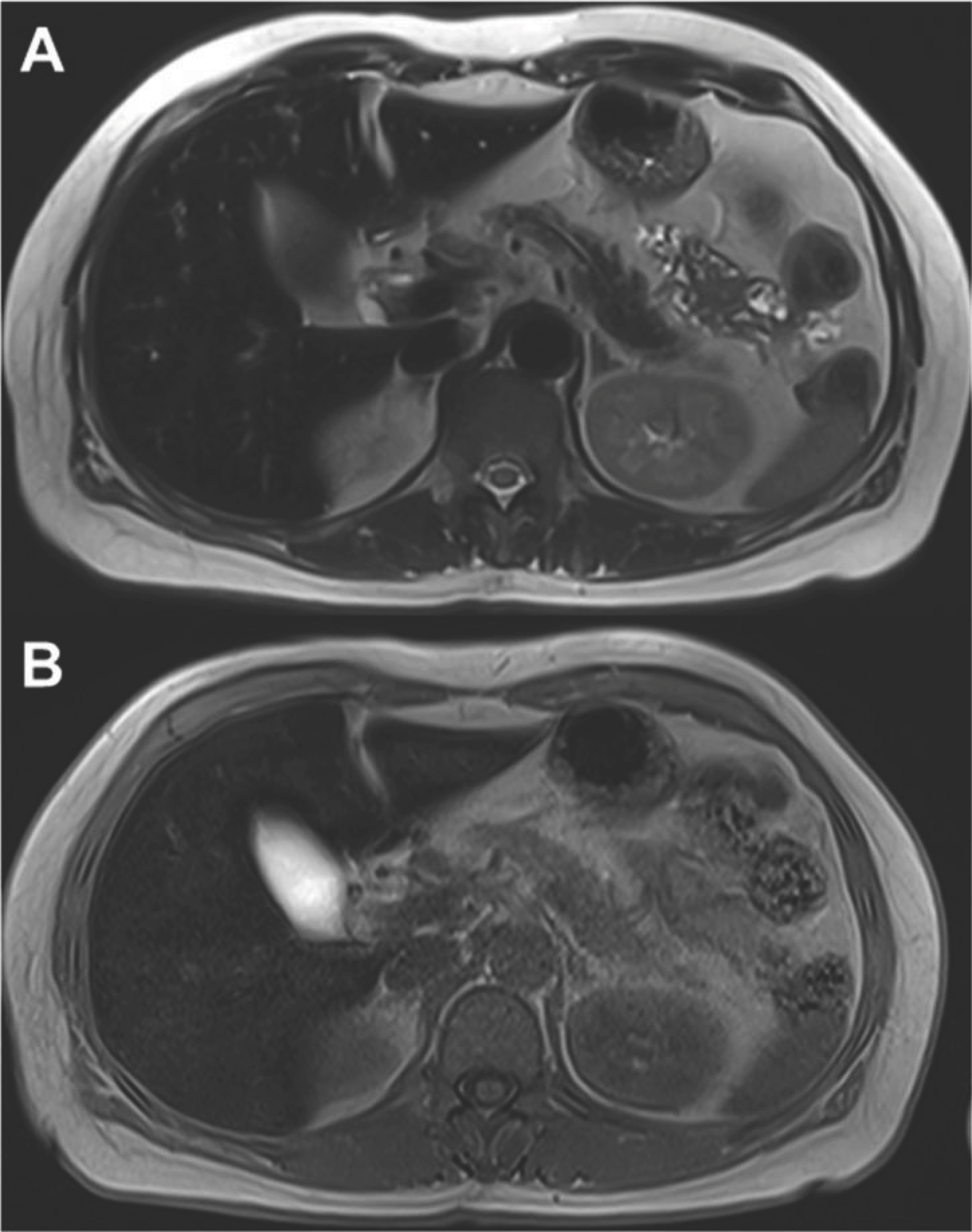
Our patient with unexplained anemia with low TSAT and high ferritin levels without inflammation was considered a high suspicion of diagnosis for some rare disorders associated with iron metabolism. Serum ferritin concentration was markedly elevated, and liver MRI revealed a low signal change of liver parenchyma representing hepatic iron deposition but the normal signal in the pancreas and bone marrow (Fig. 3). The marked iron overload seen on MRI was not compatible with hereditary hemochromatosis (HH), given the low TSAT on blood testing. A CP level was obtained, which was undetectable (< 3.0 mg/dL) (normal range, 16~45 mg/dL). We ruled out Wilson's disease because her serum copper level was reduced to 5 ╬╝ g/dL (normal range, 68~140 ╬╝ g/dL). We suspected that she might have ACP, based on typical brain MRI findings and anemia with decreased copper and CP levels. Laboratory findings are shown in Table 1. A CP gene analysis was performed at Green Cross Genome (Yongin, Korea) after the patient gave her informed consent. The identified mutation was the homozygotic substitution of the 2,630th guanine to adenine in exon 15 (NM_000096.3:c.2630 G> A), which resulted in the substitution of tryptophan for the stop codon at the 877th amino acid (p. Trp 877ŌłŚ); a disease-causing mutation that was reported in 1996 [3]. Genetic testing of the patient's siblings was not performed due to being unable to afford the associated cost.
Fig.┬Ā2.
Brain magnetic resonance imaging (MRI). (A, B) Axial T2-weighted images show bilateral and symmetrical low signal change in the putamen, globus pallidus, thalamus, substantia nigra, red nucleus, and dentate nucleus. (C, D) Axial SWI (susceptibility-weighted imaging) show definite dark signal change in the cerebral and cerebellar cortex (cortical pencil lining) in addition to above mentioned signal changed areas.
We diagnosed her with type 2 diabetes mellitus combined with ACP. Pigmentary retinopathy was not evident on screening assessment after diagnosis of ACP. We initiated her treatment with 15 mg/kg/day iron chelator, deferasirox. Serum ferritin was moni-tored every month, and the dose of deferasirox was adjusted if serum ferritin level had reached the target range (below 500 ng/mL) to minimize the risk of over chelation. In 18 months of follow-up, she was doing well with glycemic control on vildagliptin 50 mg twice daily and metformin 500 mg twice daily. There was an improvement in the HbA1c from 11.9% to 6.4% during that time. Eighteen months after iron chelating therapy, fasting C-peptide levels were measured at 1.32 ng/mL. The calculated HOMA-IR (homeostasis model assessment for insulin resistance) was 1.7, and HOMA-╬▓ (homeostasis model assessment of ╬▓-cell function) was 32. The patient is neurologically as-ymptomatic throughout the follow-up.
Fig.┬Ā3.
Abdominal magnetic resonance imaging. (A, B) Axial T1- and T2-weighted images show low signal change in liver parenchyma.
Table┬Ā1.
Laboratory findings
DISCUSSION
ACP is a rare disorder of iron metabolism resulting from mutations in the CP gene located on 3q23-q25. It was first described in 1987 [4]. In Japan's non-con-sanguineous populations, it is reported to occur in one per two million people [5]. However, its true in-cidence is unknown in Korea, and only several cases were reported [6]. Iron accumulates not because of systemic overload as the case in type 1 HH but because of reduced cellular iron egress secondary to absent CP.
Iron deposition and oxidative stress are central to the pathophysiology of the end-organ damage and clinical sequelae of ACP. Iron accumulation in the liver may be significant without evidence of structur-al damage or functional deficit. The reasons for this are not unclear; however, it is hypothesized that the high concentrations of antioxidant enzymes in the liver may be protective [7,8]. By contrast, deposition in the brain and pancreas results in progressive impairment. In the pancreas, beta cells are susceptible to oxidative stress, and minimal iron deposition has been observed to cause damage [9]. Iron deposition in the central nervous system is a distinctive feature of ACP compared with other iron metabolism disorders, such as type 1 HH. In ACP, there is an extensive and progressive iron deposition and subsequent oxidative stress within astrocytes and neurons in the basal ganglia, thalamus, and cerebral and cerebellar cortices. This pathogenesis leads to the broad, progressive phenotype of cerebellar ataxia, involuntary movements, parkinsonism, cognitive impairment, and psychiatric manifestations [5,10].
In neuroradiologic findings of ACP patients, brain CT shows abnormal high-density areas in the basal ganglia [11]. Furthermore, T2-weighted brain MRI and SWI MRI studies showed bilateral symmetric hypointensity of cerebral & cerebellar cortex, striatum, GP, thalamus, SN, red nucleus, locus ceruleus, cau-date nuclei, and dentate nucleus [12]. T2 hypointen-sities of deep nuclei, such as basal ganglia, due to deposits of iron or other paramagnetic substances are characteristic of other neurodegenerative disorders such as hemochromatosis, pantothenate ki-nase-associated neurodegeneration, formerly called Hallervorden-Spatz disease, multiple system atrophy and Wilson disease. Differentiation of these is based on the lesion site and characteristic signs [12]. In this case, on SWI, the hypointensity of distribution is su-perficial (cerebral cortex and cerebellar folia), and lesional hypointensity is striking. Therefore, SWI sug-gests more sensitive than T2-weighted MRI or other image sequences for finding ACP.
Diagnosis of ACP patients is usually based on the evidence of very low or undetectable serum CP levels and clinical, biochemical, or radiologic signs of iron overload target organs. From systematic reviews of case reports/series, patientsŌĆÖ clinical and biochemical features highlight substantial phenotype heterogeneity, contributing to the difficulties and delay in diagnosing such a rare disease [2,13].
According to a case series review, diabetes mellitus was the first manifestation of ACP in 69% of cases [14]. Diabetes mellitus in ACP had its clinical onset at age ~40 years, and its manifestation with insulin deficiency was usually in people of normal weight. Secondary diabetes mellitus in ACP is caused by a marked reduction of insulin-producing ╬▓ cells in the pancreas, despite the absence of islet cell autoanti-bodies against GAD and tyrosine phosphatase-like protein [15,16]. Problems with glycemic control could also be used as an additional clue and lead to the early consideration of ACP in neurologically asymp-tomatic cases. In our case, she was diagnosed with type 2 diabetes because of being overweight, rea-sonable glucose control with only oral hypoglycemic agents, serum normal fasting C-peptide levels, and her family history of diabetes. At first, she did not show early-onset insulin-dependent diabetes mellitus, and we did not suspect ACP. So, we ignored high attenuation in the liver of abdominal CT findings, suggesting hepatic iron overload.
However, we did not miss the unexplained anemia of our present case in association with low TSAT and paradoxically high ferritin without inflammation. This type of anemia must be differentiated from other common microcytosis causes, such as iron deficiency anemia. Ferritin levels are high, serum iron and TSAT levels are low (in contrast to disorders of iron overload, such as type 1 HH), serum copper content is low or average (in contrast to Wilson disease), and CP is uniquely absent [10]. Our present case sup-ports that unexplained anemia, often but not neces-sarily microcytic, in association with low TSAT and paradoxically high ferritin are the best indicators for an early diagnosis of ACP.
Recent studies showed that ACP phenotypes are more heterogeneous than previously believed and that some manifestations may appear 10~20 years earlier before the onset of obvious neurological signs [17]. Neurological symptoms usually appear in the fifth decade of life. However, ACP patients with brain iron deposition without or with only very mild neurological symptoms even after 50 years of age have been reported. These findings suggest that genetic and acquired factors may partially modify neurolog-ic phenotype [17]. Clinically, diabetes may precede neurological derangements even by decades. However, the mechanism of pancreatic endocrine damage secondary to iron accumulation and toxicity in ACP is not clearly explained at the cellular and molecular levels. Because intracellular iron deposition in pancreatic endocrine cells has been reported in neither ACP patients nor mice models of ACP [9,18]. Islet ╬▓ cell distress might be related to high sensitivity to iron toxicity or iron-deficient status induced by iron retention in the surrounding cells [19]. According to a case series review, around 60% of patients with diabetes require insulin treatment. However, about 10% of patients need only oral hypoglycemic agents or dietary therapy [14]. Our patient was also treated with oral hypoglycemic agents and lifestyle modification. We speculate that her family susceptibility, concomitant overweight, iron chelating therapy, and other unknown acquired factors may be involved in preserving ╬▓ cell function. In the future, we should closely monitor her ╬▓ cells function and aggravation of glycemic control requiring insulin therapy.
Iron chelation therapy is the mainstay of treatment for ACP. Both desferrioxamine (parenteral) and deferasirox (oral) reduce serum ferritin levels and visceral (hepatic) iron deposition in ACP patients treated with iron chelation [5]. In patients with ACP, the initiation of iron chelation therapy has been associated with improved glycemic control [5]. Interestingly, iron chelation therapy has also been shown to potentiate islet cell survival and function in islet transplant re-cipients [20]. Further studies are required regarding the mechanisms leading to neurological manifestation and development of diabetes and iron chelation therapy efficacy.
In summary, physicians must keep in mind that type 2 diabetes may be the clinical manifestation of ACP with unexplained atypical anemia but without neurological abnormalities. Atypical anemia, often with microcytosis, low TSAT, and high ferritin and typical radiologic findings, are the best signs for early diagnosis of ACP to avoid treatment delay and neurological damage.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print